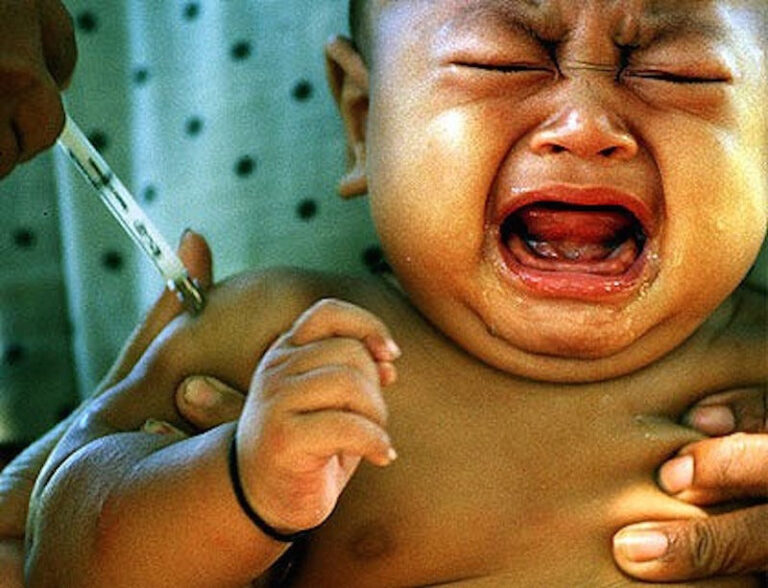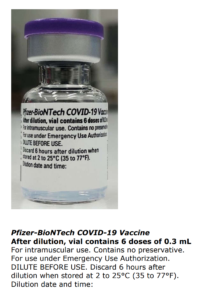
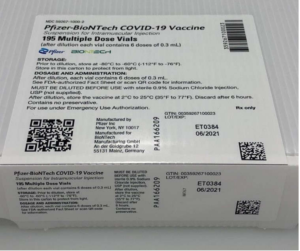
Pfizer Comirnaty tilrauna bóluefnið er á Neyðarleyfi – Stendur skýrum stöfum utan á lyfjaglasinu og á umbúðum tilraunalyfsins þann 6. júní 2021.
Updated vial and carton labels for Pfizer-BioNTech COVID-19 Vaccine with English-only labelling

SAMANTEKT Á EIGINLEIKUM LYFSINS COMIRNATY STUNGULYFSÞYKKNI ÖRDREIFA COVID-19 mRNA bóluefni með kirnisbreytingum (nucleoside modified)
- Þetta lyf er undir sérstöku eftirliti til að nýjar upplýsingar um öryggi lyfsins komist fljótt og örugglega til skila.
- Heilbrigðisstarfsmenn eru hvattir til að tilkynna allar aukaverkanir sem grunur er um að tengist lyfinu.
- Í kafla 4.8 eru upplýsingar um hvernig tilkynna á aukaverkanir.
1. HEITI LYFS
- Comirnaty stungulyfsþykkni, ördreifa
- COVID-19 mRNA bóluefni [með kirnisbreytingum (nucleoside modified)]
2. INNIHALDSLÝSING
- Lyfið er í fjölskammta hettuglasi og þarf að þynna fyrir notkun.
- Eitt hettuglas (0,45 ml) inniheldur 6 skammta sem hver er 0,3 ml eftir þynningu, sjá kafla 4.2 og 6.6.
- 1 skammtur (0,3 ml) inniheldur 30 míkrógrömm af COVID-19 mRNA bóluefni (innfellt í fitunanóagnir).
- Einþátta, mótandi RNA (mRNA) með hettu á 5’-endanum (5’-capped), framleitt með frumulausri in vitro umritun frá samsvarandi DNA sniðmátum sem kóða fyrir gaddaprótín (spike (S) protein) SARS-CoV-2 veirunnar.
- Sjá lista yfir öll hjálparefni í kafla 6.1.
3. LYFJAFORM
- Stungulyfsþykkni, ördreifa (sæft þykkni).
- Bóluefnið er hvít eða beinhvít frosin ördreifa (pH: 6,9-7,9).

4. KLÍNÍSKAR UPPLÝSINGAR
4.1 Ábendingar
- Comirnaty er ætlað til virkrar bólusetningar gegn COVID-19 af völdum SARS-CoV-2 veiru hjá einstaklingum 12 ára og eldri.
- Notkun bóluefnisins skal vera í samræmi við opinberar ráðleggingar.
4.2 SKAMMTAR OG LYFJAGJÖF
Skammtar
Einstaklingar 12 ára og eldri
- Comirnaty er gefið í vöðva eftir þynningu sem bólusetning með 2 skömmtum (0,3 ml hver).
- Ráðlagt er að gefa seinni skammtinn 3 vikum eftir að fyrri skammturinn er gefinn (sjá kafla 4.4 og 5.1).
- Engar upplýsingar liggja fyrir um hvort hægt sé að skipta Comirnaty út fyrir önnur COVID-19 bóluefni til að ljúka bólusetningunni.
- Einstaklingar sem hafa fengið 1 skammt af Comirnaty eiga að fá annan skammt af Comirnaty til að ljúka bólusetningunni.
BÖRN
- Ekki hefur enn verið sýnt fram á öryggi og verkun Comirnaty hjá börnum yngri en 12 ára.
- Takmarkaðar upplýsingar liggja fyrir.
Aldraðir
- Ekki er þörf á aðlögun skammta hjá öldruðum einstaklingum ≥ 65 ára.
Lyfjagjöf
- Comirnaty á að gefa í vöðva eftir þynningu (sjá kafla 6.6).
- Eftir þynningu innihalda hettuglös með Comirnaty sex 0,3 ml skammta af bóluefni.
- Til að ná sex skömmtum úr einu hettuglasi skal nota sprautur og/eða nálar með litlum rúmmálsleifum (dead-volume).
- Sprautan og nálin eiga samtals að hafa að hámarki 35 míkrólítra rúmmálsleifar.
- Séu venjulegar sprautur og nálar notaðar, er ekki víst að nægilegt magn verði eftir til að draga sjötta skammtinn úr einu hettuglasi.
Óháð tegund sprautu og nálar:
- Hver skammtur verður að innihalda 0,3 ml af bóluefni.
- Ef magn bóluefnisins sem eftir er í hettuglasinu nær ekki heilum 0,3 ml skammti, skal farga hettuglasinu ásamt hugsanlegu umframmagni.
- Ekki má sameina umframmagn bóluefnis úr mörgum hettuglösum.
- Æskilegur staður er axlarvöðvinn á upphandleggnum.
- Ekki má dæla bóluefninu í æð, undir húð eða í húð.
- Ekki má blanda bóluefninu saman við önnur bóluefni eða lyf í sömu sprautu.
- Varúðarráðstafanir sem þarf að gera áður en bóluefnið er gefið, sjá kafla 4.4.
- Sjá leiðbeiningar í kafla 6.6 varðandi þíðingu, meðhöndlun og förgun bóluefnisins.
4.3 FRÁBENDINGAR
- Ofnæmi fyrir virka efninu eða einhverju hjálparefnanna sem talin eru upp í kafla 6.1.
4.4 SÉRSTÖK VARNAÐARORÐ OG VARÚÐARREGLUR VIÐ NOTKUN
- Rekjanleiki
- Til þess að bæta rekjanleika líffræðilegra lyfja skal heiti og lotunúmer lyfsins sem gefið er vera skráð með skýrum hætti.
Almennar ráðleggingar
Ofnæmi og bráðaofnæmi
- Tilkynnt hefur verið um tilvik bráðaofnæmis.
- Viðeigandi læknismeðferð og -eftirlit skal ávallt vera til staðar ef bráðaofnæmisviðbrögð koma fram eftir gjöf bóluefnisins.
- Mælt er með nánu eftirliti í a.m.k. 15 mínútur eftir bólusetningu.
- Ekki má gefa þeim sem fengu bráðaofnæmi eftir fyrsta skammtinn af Comirnaty annan skammt af bóluefninu.
Hjartavöðvabólga og gollurshússbólga
- Örsjaldan hafa tilvik hjartavöðvabólgu (myocarditis) og gollurshússbólgu (pericarditis) komið fyrir eftir bólusetningu með Comirnaty.
- Þessi tilvik hafa aðallega komið fyrir innan 14 daga frá bólusetningu, oftar eftir seinni bólusetninguna og oftar hjá yngri karlmönnum.
- Fyrirliggjandi gögn benda til þess að framvinda hjartavöðvabólgu og gollurshússbólgu eftir bólusetningu sé ekki frábrugðin framvindu hjartavöðvabólgu og gollurshússbólgu almennt.
- Bólusettum einstaklingum skal leiðbeint um að leita tafarlaust til læknis ef þeir fá (bráð og þrálát) einkenni sem benda til hjartavöðvabólgu eða gollurshússbólgu, svo sem brjóstverk, mæði eða hjartsláttarónot í kjölfar bólusetningar.
- Heilbrigðisstarfsmenn skulu fylgja viðeigandi leiðbeiningum og/eða ráðfæra sig við sérfræðilækna til að greina og meðhöndla þessa sjúkdóma.
Kvíðatengd viðbrögð
- Kvíðatengd viðbrögð, þar með talin æða- og skreyjuviðbrögð (yfirlið), oföndun eða streitutengd viðbrögð t.d. sundl, hjartsláttarónot, aukin hjartsláttartíðni, breytingar á blóðþrýstingi.
- Náladofi og svitamyndun) geta komið fram í tengslum við bólusetningarferlið sjálft.
- Streitutengd viðbrögð eru skammvinn og ganga til baka af sjálfu sér.
- Ráðleggja skal einstaklingum að láta bólusetningaraðilann vita af einkennum til þess að hann geti lagt mat á þau.
- Mikilvægt er að varúðarráðstafanir séu gerðar til að koma í veg fyrir meiðsli vegna yfirliðs.
Samhliða veikindi
Fresta skal bólusetningu hjá einstaklingum með bráð veikindi með háum hita eða bráða sýkingu. Ekki þarf að fresta bólusetningu ef um er að ræða minniháttar sýkingu og/eða vægan hita.
Blóðflagnafæð og blóðstorkusjúkdómar
- Eins og við á um aðrar inndælingar í vöðva skal gæta varúðar við gjöf bóluefnisins hjá einstaklingum sem eru á meðferð með segavarnarlyfjum eða eru með blóðflagnafæð eða blóðstorkusjúkdóm (eins og dreyrasýki), vegna þess að blæðing eða mar getur komið fram eftir gjöf í vöðva hjá þessum einstaklingum.
Ónæmisbældir einstaklingar
- Verkun, öryggi og ónæmissvörun við bóluefninu hefur ekki verið metið hjá ónæmisbældum einstaklingum, þ.m.t. hjá þeim sem eru á ónæmisbælandi meðferð.
- Hugsanlegt er að verkun Comirnaty sé minni hjá ónæmisbældum einstaklingum.
Tímalengd varnar
- Ekki er þekkt hversu lengi vörn bóluefnisins varir, þar sem klínískar rannsóknir á því standa enn yfir.
- Takmarkanir á árangri af notkun bóluefnisins
- Eins og við á um öll bóluefni, er ekki víst að bólusetning með Comirnaty veiti vörn hjá öllum einstaklingum sem fá bóluefnið.
- Ekki er víst að einstaklingar fái fulla vörn fyrr en 7 dögum eftir annan skammt bóluefnisins.
Hjálparefni
- Bóluefnið inniheldur minna en 1 mmól (39 mg) af kalíum í hverjum skammti, þ.e.a.s. er sem næst kalíumlaust.
- Bóluefnið inniheldur minna en 1 mmól (23 mg) af natríum í hverjum skammti, þ.e.a.s. er sem næst natríumlaust.
4.5 Milliverkanir við önnur lyf og aðrar milliverkanir
- Ekki hafa verið gerðar neinar rannsóknir á milliverkunum.
- Samhliða gjöf Comirnaty og annarra bóluefna hefur ekki verið rannsökuð.
4.6 FRJÓSEMI, MEÐGANGA OG BRJÓSTAGJÖF
MEÐGANGA
- Takmörkuð reynsla liggur fyrir af notkun Comirnaty á meðgöngu.
- Dýrarannsóknir benda hvorki til beinna né óbeinna skaðlegra áhrifa á meðgöngu, þroska fósturvísis/fósturs, fæðingu eða þroska eftir fæðingu (sjá kafla 5.3).
- Aðeins skal íhuga gjöf Comirnaty á meðgöngu ef hugsanlegir kostir vega þyngra en hugsanleg áhætta fyrir móður og fóstur.
BRJÓSTAGJÖF
- Ekki er þekkt hvort Comirnaty skilst út í brjóstamjólk.
FRJÓSEMI
- Dýrarannsóknir benda hvorki til beinna né óbeinna skaðlegra áhrifa á æxlun (sjá kafla 5.3).
4.7 ÁHRIF Á HÆFNI TIL AKSTURS OG NOTKUNAR VÉLA
- Comirnaty hefur engin eða óveruleg áhrif á hæfni til aksturs og notkunar véla.
- Hins vegar geta sum áhrifin sem nefnd eru í kafla 4.8 dregið tímabundið úr hæfni til aksturs eða notkunar véla.
4.8 AUKAVERKANIR
Samantekt á öryggi
- Öryggi Comirnaty var metið hjá þátttakendum 12 ára og eldri í 2 klínískum rannsóknum sem tóku til 22 875 þátttakenda (21 744 þátttakendur sem voru 16 ára og eldri og 1 131 unglingar sem voru 12 til 15 ára) sem höfðu fengið a.m.k. einn skammt af Comirnaty.
- Heildaröryggi Comirnaty hjá unglingum 12 til 15 ára var svipað og hjá þátttakendum sem voru 16 ára og eldri.
Þátttakendur 16 ára og eldri
- Í Rannsókn 2 fengu alls 21 720 þátttakendur 16 ára og eldri a.m.k. 1 skammt af Comirnaty og alls 21 728 þátttakendur 16 ára og eldri fengu lyfleysu (þ.m.t. 138 unglingar á aldrinum 16 til 17 ára í bóluefnishópnum og 145 unglingar á aldrinum 16 til 17 ára í lyfleysuhópnum).
- Alls fengu 20 519 þátttakendur 16 ára og eldri 2 skammta af Comirnaty.
- Við greiningu á gögnum úr Rannsókn 2 voru alls 19 067 þátttakendur 16 ára og eldri (9 531 sem fengu Comirnaty og 9 536 sem fengu lyfleysu) metnir með tilliti til öryggis í a.m.k. 2 mánuði eftir seinni skammtinn af Comirnaty.
- Þetta tók til alls 10 727 þátttakenda 16 til 55 ára (5 350 sem fengu Comirnaty og 5 377 sem fengu lyfleysu) og alls 8 340 þátttakenda 56 ára og eldri (4 181 sem fengu Comirnaty og 4 159 sem fengu lyfleysu).
- Algengustu aukaverkanirnar hjá þátttakendum 16 ára og eldri voru verkur á stungustað (> 80%), þreyta (> 60%), höfuðverkur (> 50%), vöðvaverkir og kuldahrollur (> 30%), liðverkir (> 20%), hiti og bólga á stungustað (> 10%), voru venjulega vægar eða miðlungsmiklar og gengu til baka fáum dögum eftir bólusetningu.
- Örlítið lægri tíðni viðbragða við bóluefninu (reactogenicity) tengdist hærri aldri.
- Öryggissnið 545 þátttakenda 16 ára og eldri sem fengu Comirnaty og voru sermijákvæðir fyrir SARS-CoV-2 í upphafi rannsóknarinnar var svipað því sem sást hjá almenna þýðinu.
Unglingar 12 til 15 ára
- Í greiningu á rannsókn 2, byggt á upplýsingum sem fengnar voru allt til lokadagsetningar greiningar (cutoff date) þann 13. mars 2021, voru 2 260 unglingar (1 131 sem fengu Comirnaty og 1 129 sem fengu lyfleysu) á aldrinum 12 til 15 ára.
- Af þeim hefur 1 308 unglingum (660 sem fengu Comirnaty og 648 sem fengu lyfleysu) verið fylgt eftir í a.m.k. 2 mánuði eftir seinni skammtinn af Comirnaty.
- Öryggismat rannsóknar 2 er enn í gangi.
- Algengustu aukaverkanirnar hjá unglingum 12 til 15 ára voru verkur á stungustað (> 90%), þreyta og höfuðverkur (> 70%), vöðvaverkir og kuldahrollur (> 40%), liðverkir og hiti (> 20%).
Tafla með aukaverkunum úr klínískum rannsóknum og upplýsingum um reynslu eftir markaðssetningu hjá einstaklingum 12 ára og eldri
Aukaverkanir sem komu fram í klínískum rannsóknum eru taldar upp hér fyrir neðan samkvæmt eftirfarandi tíðniflokkun:
- Mjög algengar (≥ 1/10),
- Algengar (≥ 1/100 til <1/10),
- Sjaldgæfar (≥ 1/1 000 til < 1/100),
- Mjög sjaldgæfar (≥ 1/10 000 til < 1/1 000),
- Koma örsjaldan fyrir (< 1/10 000),
- Tíðni ekki þekkt (ekki hægt að áætla tíðni út frá fyrirliggjandi gögnum).
Tafla 1: Aukaverkanir úr klínískum rannsóknum á Comirnaty og reynsla eftir:
- Á tímabilinu eftir markaðssetningu hefur verið tilkynnt um bólgu í andliti hjá einstaklingum sem fengu bóluefni og voru með sögu um inndælingu á húðfylliefnum.
Tilkynning aukaverkana sem grunur er um að tengist lyfinu
- Eftir að lyf hefur fengið markaðsleyfi er mikilvægt að tilkynna aukaverkanir sem grunur er um að tengist því.
- Þannig er hægt að fylgjast stöðugt með sambandinu milli ávinnings og áhættu af notkun lyfsins.
- Heilbrigðisstarfsmenn eru hvattir til að tilkynna allar aukaverkanir sem grunur er um að tengist lyfinu samkvæmt fyrirkomulagi sem gildir í hverju landi fyrir sig, sjá Appendix V og láta lotunúmer fylgja ef það er til staðar.
4.9 OFSKÖMMTUN
- Upplýsingar um ofskömmtun liggja fyrir frá 52 þátttakendum í klínísku rannsókninni sem fengu 58 míkrógrömm af Comirnaty vegna rangrar þynningar.
- Þeir sem fengu bóluefnið tilkynntu ekki um aukningu á viðbrögðum (reactogenicity) eða aukaverkunum.
- Við ofskömmtun er ráðlagt að fylgjast með lífsmörkum og hugsanlega veita meðferð samkvæmt einkennum.
5. LYFJAFRÆÐILEGAR UPPLÝSINGAR
5.1 Lyfhrif
- Flokkun eftir verkun: bóluefni, önnur veirubóluefni, ATC-flokkur: J07BX03
Verkunarháttur
- mRNA með kirnisbreytingum, sem er í Comirnaty, er í fitunanóögnum sem gerir RNA sem ekki er eftirmyndandi (non-replicating) kleift að komast inn í hýsilfrumur til að stýra skammvinnri tjáningu á SARS-CoV-2 S mótefnavakanum.
- mRNA kóðar fyrir S í fullri lengd sem fest er við himnu (membrane-anchored) og er með tvær punktstökkbreytingar í miðspíralnum.
- Stökkbreyting þessara tveggja amínósýra í prólín læsir S í þrívíddarlögun áður en það binst frumum og veldur samruna veirunnar við þær, sem er ákjósanleg með tilliti til hlutverks þess sem mótefnavaka.
- Bóluefnið kallar bæði fram hlutleysandi mótefni og frumubundna ónæmissvörun við S-mótefnavakanum, sem getur stuðlað að vernd gegn COVID-19.
Verkun
- Rannsókn 2 er fjölsetra, fjölþjóða, 1/2/3. stigs, slembiröðuð, blinduð (observer blind) verkunarrannsókn með samanburði við lyfleysu hjá þátttakendum 12 ára og eldri til ákvörðunar á skömmtum bóluefnisins og til að velja bóluefni sem komu til greina.
- Slembiröðun var lagskipt eftir aldri: 12 til 15 ára, 16 til 55 ára og 56 ára og eldri, þar sem að lágmarki 40% þátttakenda voru í ≥ 56 ára laginu.
- Rannsóknin útilokaði þátttakendur með ónæmisskerðingu og þá sem höfðu áður fengið klíníska eða örverufræðilega greiningu á COVID-19.
- Þátttakendur með stöðugan sjúkdóm sem skilgreindur var sem sjúkdómur sem krafðist ekki verulegra breytinga á meðferð eða sjúkrahúsvistunar vegna versnandi sjúkdóms síðustu 6 vikurnar fyrir skráningu fengu að taka þátt, sem og þátttakendur með þekkta stöðuga sýkingu af völdum HIV-veiru, lifrarbólguveiru C (HCV) eða lifrarbólguveiru B (HBV).
Verkun hjá þátttakendum 16 ára og eldri
- Í 2/3. stigs hlutanum í rannsókn 2, byggt á upplýsingum sem safnað var til og með 14. nóvember 2020, var u.þ.b. 44 000 þátttakendum slembiraðað jafnt til að fá 2 skammta af COVID-19 mRNA bóluefni eða lyfleysu, með 21 dags millibili.
- Verkunargreiningarnar tóku til þátttakenda sem fengu síðari bólusetninguna innan 19 til 42 daga eftir fyrri bólusetninguna.
- Flestir (93,1%) fengu seinni skammtinn 19 dögum til 23 dögum eftir skammt 1.
- Fyrirhugað er að fylgja þátttakendum eftir í allt að 24 mánuði eftir skammt 2, til að meta öryggi og verkun gegn COVID-19.
- Í klínísku rannsókninni þurftu a.m.k. 14 dagar að líða milli þess að þátttakendur fengu inflúensubóluefni og að þeir fengu annaðhvort lyfleysu eða COVID-19 mRNA bóluefni, hvort sem gerðist fyrr.
- Í klínísku rannsókninni þurftu a.m.k. 60 dagar að líða milli þess að þátttakendur fengu blóð-/plasmaafurðir eða ónæmisglóbúlín og að þeir fengu annaðhvort lyfleysu eða COVID-19 mRNA bóluefni, hvort sem gerðist fyrr, fram að lokum rannsóknarinnar.
- Þýðið til greiningar á aðalendapunkti verkunar tók til 36 621 þátttakanda 12 ára og eldri (18 242 í COVID-19 mRNA bóluefnishópnum og 18 379 í lyfleysuhópnum) sem sýndu ekki vísbendingar um fyrri sýkingu af völdum SARS-CoV-2 allt að 7 dögum eftir seinni skammtinn.
- Auk þess voru 134 þátttakendur á aldrinum 16 til 17 ára (66 í COVID-19 mRNA bóluefnishópnum og 68 í lyfleysuhópnum) og 1 616 þátttakendur 75 ára og eldri (804 í COVID-19 mRNA bóluefnishópnum og 812 í lyfleysuhópnum).
- Við greiningu á meginverkuninni hafði þátttakendum verið fylgt eftir með tilliti til COVID-19 með einkennum í alls 2 214 mannár í hópnum sem fékk COVID-19 mRNA bóluefnið og í alls 2 222 mannár í lyfleysuhópnum.
- Enginn klínískur munur sem skipti máli var á heildarverkun bóluefnisins hjá þátttakendum sem voru í hættu á að fá alvarlegan COVID-19, þar á meðal hjá þeim sem voru með 1 eða fleiri samverkandi sjúkdóma sem juku hættuna á alvarlegum COVID-19 (t.d. astma, líkamsþyngdarstuðul (BMI) ≥ 30 kg/m2, langvinnan lungnasjúkdóm, sykursýki, háþrýsting).
Upplýsingar um verkun bóluefnisins koma fram í töflu 2.
|
Tafla 2: Verkun bóluefnisins – Fyrsta COVID-19 tilfelli frá 7 dögum eftir skammt 2, eftir undirhópi aldurs – þátttakendur án vísbendinga um sýkingu innan 7 daga eftir skammt 2 – þýði með metanlega verkun (7 dagar) Fyrsta COVID-19 tilfelli frá 7 dögum eftir skammt 2 hjá þátttakendum án vísbendinga um fyrri SARS-CoV-2 sýkingu* |
|||
|
Undirhópur |
COVID-19 mRNA bóluefni Na = 18 198 tilfelli n1b Eftirlitstímic (n2d) |
Lyfleysa Na = 18 325 tilfelli n1b Eftirlitstímic (n2d) |
Verkun bóluefnis % (95% CI)e |
|
Allir þátttakendur |
8 2,214 (17 411) |
162 2,222 (17 511) |
95,0 (90,0; 97,9) |
|
16 til 64 ára |
7 1,706 (13 549) |
143 1,710 (13 618) |
95,1 (89,6; 98,1) |
|
65 ára og eldri |
1 0,508 (3848) |
19 0,511 (3880) |
94,7 (66,7; 99,9) |
|
65 til 74 ára |
1 0,406 (3074) |
14 0,406 (3095) |
92,9 (53,1; 99,8) |
|
75 ára og eldri |
0 0,102 (774) |
5 0,106 (785) |
100,0 (-13,1; 100,0) |
Athugið:
Staðfest tilfelli voru ákvörðuð með kjarnsýrumögnun (Reverse Transcription-Polymerase Chain Reaction, RT-PCR) og að minnsta kosti 1 einkenni sem samræmdist COVID-19
[*Skilgreining á tilfelli: (að minnsta kosti 1 af) hiti, nýtilkominn eða versnandi hósti, nýtilkomin eða versnandi mæði, kuldahrollur, nýtilkomnir eða versnandi vöðvaverkir, nýtilkomið tap á bragð- eða lyktarskyni, hálsbólga, niðurgangur eða uppköst.]
* Þátttakendur án sermis- eða veirufræðilegra vísbendinga (innan 7 daga eftir síðasta skammtinn) um fyrri SARS-CoV-2 sýkingu (þ.e. engin N-bindandi mótefni [sermi] í heimsókn 1 og SARS-CoV-2 greindist ekki með kjarnsýrumögnun (NAAT) [nefstrok] í heimsóknum 1 og 2) og sem höfðu neikvætt NAAT (nefstrok) í öllum óskipulögðum heimsóknum innan 7 daga eftir skammt 2 voru teknir með í greininguna.
- a. N = Fjöldi þátttakenda í tilgreinda hópnum.
- b. n1 = Fjöldi þátttakenda sem uppfylltu skilgreiningu endapunktsins.
- c. Heildareftirlitstími í 1000 mannár fyrir tiltekinn endapunkt hjá öllum þátttakendum innan hvers hóps í hættu fyrir endapunktinn. Tímabil fyrir uppsöfnun COVID-19 tilfella er frá 7 dögum eftir skammt 2 til loka eftirlitstímabilsins.
- d. n2 = Fjöldi þátttakenda í hættu fyrir endapunktinn.
- e. Öryggisbil (CI) fyrir verkun bóluefnis er byggt á Clopper og Pearson aðferðinni og aðlagað að eftirlitstímanum. CI var ekki leiðrétt fyrir margfeldni (multiplicity).
- Verkun COVID-19 mRNA bóluefnis til að koma í veg fyrir fyrsta COVID-19 tilvik frá degi 7 eða seinna eftir skammt 2 var 94,6% (95% öryggisbil 89,6% til 97,6%) samanborið við lyfleysu hjá þátttakendum 16 ára og eldri með eða án vísbendinga um fyrri sýkingu af völdum SARS-CoV-2.
- Að auki sýndu greiningar á aðalendapunkti verkunar hjá undirhópum sambærilegt punktmat á verkun óháð kyni, þjóðaruppruna og hjá þátttakendum með samverkandi sjúkdóma sem tengjast mikilli hættu á alvarlegum COVID-19.
- Verkun og ónæmissvörun hjá unglingum 12 til 15 ára
- Í greiningu á rannsókn 2 hjá unglingum á aldrinum 12 til 15 ára án vísbendinga um fyrri sýkingu komu engin tilvik fram hjá 1 005 þátttakendum sem fengu bóluefnið en 16 tilvik komu fram hjá þeim 978 sem fengu lyfleysu.
- Punktmat varðandi verkun er 100% (95% öryggisbil 75,3; 100,0).
- Hjá þátttakendum með eða án vísbendinga um fyrri sýkingu komu 0 tilvik fram hjá þeim 1 119 sem fengu bóluefnið og 18 tilvik hjá þeim 1 110 þátttakendum sem fengu lyfleysu.
- Þetta gefur einnig til kynna að punktmat varðandi verkun sé 100% (95% öryggisbil 78,1; 100,0).
- Í rannsókn 2 var greining á SARS-CoV-2 hlutleysandi títrum 1 mánuði eftir skammt 2 framkvæmd hjá undirhópi þátttakenda sem valinn var með slembiröðun og var án sermis- eða veirufræðilegra vísbendinga um fyrri SARS-CoV-2 sýkingu allt að 1 mánuði eftir skammt 2, þar sem svörun var borin saman hjá unglingum 12 til 15 ára (n = 190) og þátttakendum 16 til 25 ára (n = 170).
- Hlutfall margfeldismeðaltala títra (GMT) hjá aldurshópnum 12 til 15 ára samanborið við aldurshópinn 16 til 25 ára var 1,76, með 2-hliða 95% CI sem nam 1,47 til 2,10. Því var 1,5-földu viðmiði um „ekki lakari“-verkun náð þar sem lægri mörk 2-hliða 95% CI hvað varðar hlutfall margfeldismeðaltala [GMR] var > 0,67.
BÖRN
- Lyfjastofnun Evrópu hefur frestað kröfu um að lagðar séu fram niðurstöður úr rannsóknum á Comirnaty hjá börnum til að koma í veg fyrir COVID-19 (sjá upplýsingar í kafla 4.2 um notkun handa börnum).
- Þetta lyf hefur fengið markaðsleyfi með svokölluðu „skilyrtu samþykki“.
- Það þýðir að beðið er eftir frekari gögnum um lyfið.
- Lyfjastofnun Evrópu metur nýjar upplýsingar um lyfið að minnsta kosti árlega og uppfærir samantekt á eiginleikum lyfsins eftir því sem þörf krefur.
5.2 LYFJAHVÖRF
- Á ekki við.
5.3 FORKLÍNÍSKAR UPPLÝSINGAR
Forklínískar upplýsingar benda ekki til neinnar sérstakrar hættu fyrir menn, á grundvelli hefðbundinna rannsókna á eiturverkunum eftir endurtekna skammta og eiturverkunum á æxlun og þroska.
ALMENNAR EITURVERKANIR
- Rottur sem fengu Comirnaty í vöðva (3 heila skammta handa mönnum einu sinni í viku sem mynda tiltölulega hærri þéttni hjá rottum vegna mismunar á líkamsþyngd) sýndu nokkurn bjúg og roða á stungustað og fjölgun hvítra blóðkorna (þ.m.t. basakyrninga (basophils) og eósínfíkla), sem samræmist bólgusvörun, ásamt frymisbólumyndun í lifrarfrumum í lifrarporti án vísbendinga um lifrarskaða.
- Öll áhrifin voru afturkræf.
Eiturverkanir á erfðaefni/krabbameinsvaldandi áhrif
- Hvorki voru gerðar rannsóknir á eiturverkunum á erfðaefni né krabbameinsvaldandi áhrifum.
- Ekki er gert ráð fyrir að efnisþættir bóluefnisins (fituefni og mRNA) hafi eiturverkanir á erfðaefni.
Eiturverkanir á æxlun
- Eiturverkanir á æxlun og þroska voru rannsakaðar hjá rottum í samsettri rannsókn á frjósemi og eiturverkunum á þroska þar sem kvenkyns rottum var gefið Comirnaty í vöðva fyrir mökun og á meðgöngu (4 heila skammta handa mönnum sem mynda tiltölulega hærri þéttni hjá rottum vegna mismunar á líkamsþyngd allt frá degi 21 fyrir mökun og að meðgöngudegi 20).
- Svörun með SARS-CoV-2 hlutleysandi mótefnum var til staðar hjá móður frá því fyrir mökun og allt til loka rannsóknarinnar á degi 21 eftir fæðingu, sem og hjá fóstrum og afkvæmum.
- Engin áhrif sem tengdust bóluefninu komu fram á frjósemi kvendýra, meðgöngu eða þroska fósturvísis, fósturs eða afkvæmis.
- Engar upplýsingar liggja fyrir um Comirnaty hvað varðar flutning bóluefnisins yfir fylgju eða útskilnað í mjólk.
6. LYFJAGERÐARFRÆÐILEGAR UPPLÝSINGAR
6.1 Hjálparefni
- ((4-hýdroxýbútýl)azanedíýl)bis(hexan-6,1-díýl)bis(2-hexýldekanóat) (ALC-0315)
- 2-[(pólýetýlen glýkól)-2000]-N,N-dítetradekýlasetamíð (ALC-0159)
- 1,2-Dísteróýl-sn-glýseró-3-fosfókólín (DSPC)
- Kólesteról
- Kalíumklóríð
- Kalíumtvívetnisfosfat
- Natríumklóríð
- Tvínatríumfosfat tvíhýdrat
- Súkrósi
- Vatn fyrir stungulyf
6.2 ÓSAMRÝMANLEIKI
- Ekki má blanda þessu lyfi saman við önnur lyf en þau sem nefnd eru í kafla 6.6.
6.3 GEYMSLUÞOL
- Óopnað hettuglas
- Frosið hettuglas
- 6 mánuðir við -90°C til -60°C
- Meðan á 6 mánaða geymslutíma stendur má geyma og flytja óopnuð hettuglös við -25°C til -15°C á samfelldu tímabili í allt að 2 vikur og geyma svo aftur við -90ºC til -60ºC.
- Þítt hettuglas
- 1 mánuður við 2°C til 8°C
- Af þessum 1 mánaðar geymslutíma við 2°C til 8°C má nota allt að 12 klukkustundir til að flytja bóluefnið.
- Fyrir notkun má geyma óopnað hettuglas í allt að 2 klukkustundir við hitastig allt að 30°C.
Ekki má frysta bóluefnið aftur eftir þíðingu.
- Umsjón með hitafrávikum þegar bóluefnið er fjarlægt úr frysti
- Gögn um stöðugleika gefa til kynna að óopnað hettuglas haldi stöðugleika sínum í allt að:
- 24 klst. þegar það er geymt við hitastig sem nemur frá -3°C til 2°C
- alls 4 klst. þegar það er geymt við hitastig sem nemur frá 8°C til 30°C; þ.m.t. 2 klst. við allt að 30°C eins og lýst er hér að ofan
- Þessar upplýsingar eru aðeins ætlaðar sem leiðbeiningar fyrir heilbrigðisstarfsmenn ef tímabundin hitafrávik eiga sér stað.
- Flutningur frosinna hettuglasa sem geymd eru við ofurlágt hitastig (< -60°C)
- Eftir að lokaðir bakkar sem innihalda 195 hettuglös hafa verið fjarlægðir úr frystigeymslu með ofurlágu hitastigi (< -60°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 5 mínútur.
- Eftir að opnir bakkar eða bakkar sem innihalda færri en 195 hettuglös hafa verið fjarlægðir úr frystigeymslu með ofurlágu hitastigi (< -60°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 3 mínútur.
- Eftir að bökkum með hettuglösum hefur aftur verið komið fyrir í frystigeymslu eftir að hafa verið við hita sem nemur allt að 25°C, þurfa þeir að vera í frystigeymslunni í að minnsta kosti 2 klukkustundir áður en hægt er að fjarlægja þá aftur.
- Flutningur frosinna hettuglasa sem geymd eru við -25°C til -15°C
- Eftir að lokaðir bakkar sem innihalda 195 hettuglös hafa verið fjarlægðir úr frystigeymslu (-25°C til -15°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 3 mínútur.
- Eftir að opnir bakkar eða bakkar sem innihalda færri en 195 hettuglös hafa verið fjarlægðir úr frystigeymslu (-25°C til -15°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 1 mínútu.
- Eftir að hettuglas er fjarlægt úr bakkanum skal þíða það fyrir notkun.
Þynnt lyf
- Sýnt hefur verið fram á efna- og eðlisfræðilegan stöðugleika, þ.m.t. meðan á flutningi stendur, við notkun í 6 klukkustundir við 2°C til 30°C eftir þynningu með natríumklóríð 9 mg/ml (0,9%) stungulyfi, lausn.
- Frá örverufræðilegu sjónarmiði skal nota lyfið tafarlaust, nema þynningaraðferðin útiloki hættu á örverumengun.
- Ef það er ekki notað tafarlaust eru geymslutími og geymsluskilyrði við notkun á ábyrgð notandans.
6.4 SÉRSTAKAR VARÚÐARREGLUR VIÐ GEYMSLU
- Geymið í frysti við -90°C til -60°C.
- Geymið í upprunalegum umbúðum til varnar gegn ljósi.
- Við geymslu skal lágmarka útsetningu fyrir herbergisljósi og forðast útsetningu fyrir beinu sólarljósi og útfjólubláu ljósi.
- Þídd hettuglös má meðhöndla við herbergisljós.
- Geymsluskilyrði eftir þíðingu og þynningu lyfsins, sjá kafla 6.3.
6.5 GERÐ ÍLÁTS OG INNIHALD
- 2 ml glært fjölskammta hettuglas (gler af gerð I) með tappa (brómóbútýl gervigúmmí) og smelluloki úr plasti með álinnsigli.
- Hvert hettuglas inniheldur 6 skammta, sjá kafla 6.6.
- Pakkningastærð: 195 hettuglös
6.6 SÉRSTAKAR VARÚÐARRÁÐSTAFANIR VIÐ FÖRGUN OG ÖNNUR MEÐHÖNDLUN
- Leiðbeiningar um meðhöndlun
7. MARKAÐSLEYFISHAFI
- BioNTech Manufacturing GmbH
- An der Goldgrube 12
- 55131 Mainz
- Þýskaland
- sími: +49 6131 9084-0
- bréfasími: +49 6131 9084-2121
- service@biontech.de
8. MARKAÐSLEYFISNÚMER
- EU/1/20/1528/001
9. DAGSETNING FYRSTU ÚTGÁFU MARKAÐSLEYFIS / ENDURNÝJUNAR MARKAÐSLEYFIS
- Dagsetning fyrstu útgáfu markaðsleyfis: 21. desember 2020
10. DAGSETNING ENDURSKOÐUNAR TEXTANS
- Ítarlegar upplýsingar um lyfið eru birtar á vef Lyfjastofnunar Evrópu www.ema.europa.eu.
VIÐAUKI II
- A. FRAMLEIÐENDUR LÍFFRÆÐILEGRA VIRKRA EFNA OG FRAMLEIÐENDUR SEM ERU ÁBYRGIR FYRIR LOKASAMÞYKKT
- B. FORSENDUR FYRIR, EÐA TAKMARKANIR Á, AFGREIÐSLU OG NOTKUN
- C. AÐRAR FORSENDUR OG SKILYRÐI MARKAÐSLEYFIS
- D. FORSENDUR EÐA TAKMARKANIR ER VARÐA ÖRYGGI OG VERKUN VIÐ NOTKUN LYFSINS
- E. SÉRSTÖK SKYLDA TIL AÐ LJÚKA AÐGERÐUM EFTIR ÚTGÁFU SKILYRTS MARKAÐSLEYFIS
- A. FRAMLEIÐENDUR LÍFFRÆÐILEGRA VIRKRA EFNA OG FRAMLEIÐENDUR SEM ERU ÁBYRGIR FYRIR LOKASAMÞYKKT
Heiti og heimilisfang framleiðenda líffræðilegra virkra efna
BioNTech Manufacturing GmbH
- An der Goldgrube 12
- 55131 Mainz
- Þýskaland
- BioNTech Manufacturing Marburg GmbH
- Emil-von-Behring-Strasse 76
- 35401 Marburg
- Þýskaland
Rentschler Biopharma SE
- Erwin-Rentschler-Strasse 21
- 88471 Laupheim
- Þýskaland
Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC
- 1 Burtt Road
- Andover, MA 01810
- Bandaríkin
Heiti og heimilisfang framleiðenda sem eru ábyrgir fyrir lokasamþykkt
BioNTech Manufacturing GmbH
- Kupferbergterrasse 17 – 19
- 55116 Mainz
- Þýskaland
Pfizer Manufacturing Belgium NV
- Rijksweg 12
- 2870 Puurs
- Belgía
Heiti og heimilisfang framleiðanda sem er ábyrgur fyrir lokasamþykkt viðkomandi lotu skal koma fram í prentuðum fylgiseðli.
- Í ljósi hins yfirlýsta alþjóðlega neyðarástands hvað varðar lýðheilsu og til að tryggja snemmbúna afhendingu, er lyfið háð tímabundinni undanþágu
- UNDANÞÁGA sem gerir það að verkum að hægt er að reiða sig á eftirlitsprófanir á lotum lyfsins sem framkvæmdar eru á skráðum setrum sem staðsett eru í þriðja landi.
- Þessi undanþága fellur úr gildi 31. ágúst 2021.
- Innleiðing aðgerða til eftirlits með lotum lyfsins innan ESB, þar með talið nauðsynlegra breytinga á skilmálum markaðsleyfisins,
- Því þarf að vera lokið í síðasta lagi 31. ágúst 2021 í samræmi við samþykkta áætlun fyrir þessa tilfærslu á prófunum
- Framvinduskýrslum þarf að skila þann 31. mars 2021 og einnig með umsókn um árlega endurnýjun.
B. FORSENDUR FYRIR, EÐA TAKMARKANIR Á, AFGREIÐSLU OG NOTKUN
- Lyfið er lyfseðilsskylt.
- Opinber lokasamþykkt
- Samkvæmt ákvæðum 114. greinar í tilskipun 2001/83/EB annast opinber rannsóknarstofa eða rannsóknarstofa sem tilnefnd er til þess, opinbera lokasamþykkt.
C. AÐRAR FORSENDUR OG SKILYRÐI MARKAÐSLEYFIS
Samantektir um öryggi lyfsins (PSUR)
- Skilyrði um hvernig leggja skal fram samantektir um öryggi lyfsins koma fram í lista yfir viðmiðunardagsetningar Evrópusambandsins (EURD lista) sem gerð er krafa um í grein 107c(7) í tilskipun 2001/83/EB og öllum síðari uppfærslum sem birtar eru í evrópsku lyfjavefgáttinni.
- Markaðsleyfishafi skal leggja fram fyrstu samantektina um öryggi lyfsins innan 6 mánaða frá útgáfu markaðsleyfis.
D. FORSENDUR EÐA TAKMARKANIR ER VARÐA ÖRYGGI OG VERKUN VIÐ NOTKUN LYFSINS
- Áætlun um áhættustjórnun
- Markaðsleyfishafi skal sinna lyfjagátaraðgerðum sem krafist er, sem og öðrum ráðstöfunum eins og fram kemur í áætlun um áhættustjórnun í kafla 1.8.2 í markaðsleyfinu og öllum uppfærslum á áætlun um áhættustjórnun sem ákveðnar verða.
- Leggja skal fram uppfærða áætlun um áhættustjórnun:
- Að beiðni Lyfjastofnunar Evrópu.
- Þegar áhættustjórnunarkerfinu er breytt, sérstaklega ef það gerist í kjölfar þess að nýjar upplýsingar berast sem geta leitt til mikilvægra breytinga á hlutfalli ávinnings/áhættu eða vegna þess að mikilvægur áfangi (tengdur lyfjagát eða lágmörkun áhættu) næst.
E. SÉRSTÖK SKYLDA TIL AÐ LJÚKA AÐGERÐUM EFTIR ÚTGÁFU SKILYRTS MARKAÐSLEYFIS
|
Þetta lyf hefur fengið markaðsleyfi með skilyrtu samþykki og í samræmi við grein 14-a í reglugerð (EB) nr. 726/2004 skal markaðsleyfishafi ljúka eftirfarandi innan tilgreindra tímamarka: Lýsing |
Tímamörk |
|
Til að ljúka lýsingu á eiginleikum virka efnisins og fullbúna lyfsins skal markaðsleyfishafi leggja fram viðbótargögn. |
Júlí 2021. Bráðabirgða-skýrslur: 31. mars 2021 |
|
Til að tryggja samfelld gæði lyfsins skal markaðsleyfishafi veita viðbótarupplýsingar til að bæta eftirlitsaðferðina, þ.m.t. gæðalýsingu á virka efninu og fullbúna lyfinu. |
Júlí 2021. Bráðabirgða-skýrslur: Mars 2021 |
|
Til að staðfesta hreinleika og tryggja alhliða gæðaeftirlit og samræmi á milli lota á öllum framleiðslustigum (lifecycle) fullbúna lyfsins, skal markaðsleyfishafi veita frekari upplýsingar um framleiðsluferlið (synthetic process) og eftirlitsaðferð fyrir hjálparefnið ALC-0315. |
Júlí 2021. Bráðabirgða-skýrslur: Janúar 2021, apríl 2021 |
|
Til að staðfesta hreinleika og tryggja alhliða gæðaeftirlit og samræmi á milli lota á öllum framleiðslustigum fullbúna lyfsins, skal markaðsleyfishafi veita frekari upplýsingar um framleiðsluferlið og eftirlitsaðferð fyrir hjálparefnið ALC-0159. |
Júlí 2021. Bráðabirgða-skýrslur: Janúar 2021, apríl 2021 |
|
Lýsing |
Tímamörk |
|
Til að staðfesta verkun og öryggi Comirnaty skal markaðsleyfishafi leggja fram lokaskýrslu klínískra rannsókna fyrir slembiröðuðu, blinduðu (observer blind) rannsóknina C4591001 með samanburði við lyfleysu. |
Desember 2023 |
A. ÁLETRANIR
- UPPLÝSINGAR SEM EIGA AÐ KOMA FRAM Á YTRI UMBÚÐUM
- MERKIMIÐI Á ÖSKJU
1. HEITI LYFS
- COMIRNATY stungulyfsþykkni, ördreifa COVID-19 mRNA bóluefni (með kirnisbreytingum)
2. VIRK(T) EFNI
- Eftir þynningu inniheldur hvert hettuglas 6 skammta sem hver er 0,3 ml.
3. HJÁLPAREFNI
- Hjálparefni: ALC-0315, ALC-0159, DSPC, kólesteról, kalíumklóríð, kalíumtvívetnisfosfat, natríumklóríð, tvínatríumfosfat tvíhýdrat, súkrósi, vatn fyrir stungulyf
4. LYFJAFORM OG INNIHALD
- Stungulyfsþykkni, ördreifa
- 195 fjölskammta hettuglös
5. AÐFERÐ VIÐ LYFJAGJÖF OG ÍKOMULEIÐ(IR)
- Til notkunar í vöðva eftir þynningu.
- Lesið fylgiseðilinn fyrir notkun.
- Skannið QR-kóðann til að fá frekari upplýsingar.
- Þynnið fyrir notkun: Þynnið hvert hettuglas með 1,8 ml af natríumklóríð 9 mg/ml (0,9%) stungulyfi, lausn.
6. SÉRSTÖK VARNAÐARORÐ UM AÐ LYFIÐ SKULI GEYMT ÞAR SEM BÖRN HVORKI NÁ TIL NÉ SJÁ
- Geymið þar sem börn hvorki ná til né sjá.
7. ÖNNUR SÉRSTÖK VARNAÐARORÐ, EF MEÐ ÞARF
8. FYRNINGARDAGSETNING
- EXP
- Fyrningardagsetning við 2°C til 8°C: ……………..
- (að hámarki 1 mánuður; eldri fyrningardagsetning skal gerð ólæsileg)
9. SÉRSTÖK GEYMSLUSKILYRÐI
Geymsla:
- Fyrir þynningu, geymið við -90°C til -60°C í upprunalegum umbúðum til varnar gegn ljósi.
- Eftir þynningu, geymið bóluefnið við 2°C til 30°C og notið innan 6 klukkustunda.
- Fargið öllu ónotuðu bóluefni.
10. SÉRSTAKAR VARÚÐARRÁÐSTAFANIR VIÐ FÖRGUN LYFJALEIFA EÐA ÚRGANGS VEGNA LYFSINS ÞAR SEM VIÐ Á
11. NAFN OG HEIMILISFANG MARKAÐSLEYFISHAFA
- BioNTech Manufacturing GmbH
- An der Goldgrube 12
- 55131 Mainz, Þýskaland
12. MARKAÐSLEYFISNÚMER
- EU/1/20/1528/001
13. LOTUNÚMER
- LOT
14. AFGREIÐSLUTILHÖGUN
15. NOTKUNARLEIÐBEININGAR
16. UPPLÝSINGAR MEÐ BLINDRALETRI
- Fallist hefur verið á rök fyrir undanþágu frá kröfu um blindraletur.
17. EINKVÆMT AUÐKENNI – TVÍVÍTT STRIKAMERKI
- Á pakkningunni er tvívítt strikamerki með einkvæmu auðkenni.
18. EINKVÆMT AUÐKENNI – UPPLÝSINGAR SEM FÓLK GETUR LESIÐ
- PC
- SN
- NN
- LÁGMARKS UPPLÝSINGAR SEM SKULU KOMA FRAM Á INNRI UMBÚÐUM LÍTILLA EININGA
- MERKIMIÐI Á HETTUGLASI
1. HEITI LYFS OG ÍKOMULEIÐ(IR)
- COMIRNATY sæft þykkni
- COVID-19 mRNA bóluefni
- i.m.
2. AÐFERÐ VIÐ LYFJAGJÖF
3. FYRNINGARDAGSETNING
- EXP
4. LOTUNÚMER
- LOT
5. INNIHALD TILGREINT SEM ÞYNGD, RÚMMÁL EÐA FJÖLDI EININGA
- 6 skammtar eftir þynningu
6. ANNAÐ
- Fargið (dagsetning/tími):
B. FYLGISEÐILL
- Fylgiseðill: Upplýsingar fyrir notanda lyfsins
- Comirnaty stungulyfsþykkni, ördreifa
- COVID-19 mRNA bóluefni (með kirnisbreytingum)
- Þetta lyf er undir sérstöku eftirliti til að nýjar upplýsingar um öryggi lyfsins komist fljótt og örugglega til skila.
- Allir geta hjálpað til við þetta með því að tilkynna aukaverkanir sem koma fram.
- Aftast í kafla 4 eru upplýsingar um hvernig tilkynna á aukaverkanir.
- Lestu allan fylgiseðilinn vandlega áður en þú færð bóluefnið. Í honum eru mikilvægar upplýsingar.
- Geymið fylgiseðilinn. Nauðsynlegt getur verið að lesa hann síðar.
- Leitið til læknisins, lyfjafræðings eða hjúkrunarfræðingsins ef þörf er á frekari upplýsingum.
- Látið lækninn, lyfjafræðing eða hjúkrunarfræðinginn vita um allar aukaverkanir.
- Þetta gildir einnig um aukaverkanir sem ekki er minnst á í þessum fylgiseðli. Sjá kafla 4.
Í fylgiseðlinum eru eftirfarandi kaflar
1. Upplýsingar um Comirnaty og við hverju það er notað
2. Áður en þú færð Comirnaty
3. Hvernig Comirnaty er gefið
4. Hugsanlegar aukaverkanir
5. Hvernig geyma á Comirnaty
6. Pakkningar og aðrar upplýsingar
- 1. Upplýsingar um Comirnaty og við hverju það er notað
- Comirnaty er bóluefni sem notað er til að koma í veg fyrir COVID-19 af völdum SARS-CoV-2 veiru.
- Comirnaty er gefið fullorðnum og unglingum, 12 ára og eldri.
- Bóluefnið fær ónæmiskerfið (náttúrulegar varnir líkamans) til að framleiða mótefni og blóðfrumur sem vinna gegn veirunni og veitir þannig vörn gegn COVID-19.
- Þú getur ekki fengið COVID-19 af Comirnaty af því að það inniheldur ekki veiru til myndunar ónæmis.
2. Áður en þú færð Comirnaty
Ekki má gefa Comirnaty
- Ef um er að ræða ofnæmi fyrir virka efninu eða einhverju öðru innihaldsefni lyfsins (talin upp í kafla 6).
- Varnaðarorð og varúðarreglur
Leitaðu ráða hjá lækninum, lyfjafræðingi eða hjúkrunarfræðingnum áður en þér er gefið bóluefnið ef:
- þú hefur áður fengið alvarleg ofnæmisviðbrögð eða öndunarerfiðleika eftir inndælingu á öðru bóluefni eða eftir að þér var gefið Comirnaty.
- þú finnur fyrir kvíða vegna bólusetningarferlisins eða hefur fallið í yfirlið eftir að hafa fengið inndælingu með nál.
- þú ert með alvarleg veikindi eða sýkingu með háum hita. Hins vegar máttu fá bólusetninguna ef þú ert með vægan hita eða sýkingu í efri hluta öndunarvegar eins og kvef.
- þú ert með blæðingakvilla, þú færð auðveldlega mar eða þú notar lyf til að hindra blóðtappa.
- þú ert með veiklað ónæmiskerfi vegna sjúkdóms eins og HIV-sýkingar eða lyfs eins og barkstera, sem hefur áhrif á ónæmiskerfið.
- Örsjaldan hefur verið greint frá tilvikum um hjartavöðvabólgu og gollurshússbólgu (bólga) innan tveggja vikna frá bólusetningu, oftar eftir seinni bólusetninguna og oftar hjá yngri karlmönnum.
- Eftir bólusetninguna skaltu vera vakandi fyrir einkennum hjartavöðvabólgu og gollurshússbólgu, svo sem mæði, hjartsláttarónotum og brjóstverk, og leita tafarlaust til læknis ef þú færð þessi einkenni.
- Eins og við á um öll bóluefni, þá er ekki víst að 2 skammta bólusetningin með Comirnaty veiti öllum sem hana fá fulla vörn og ekki er þekkt hversu lengi vörnin varir.
- Börn
Comirnaty er ekki ætlað börnum yngri en 12 ára.
- Notkun annarra lyfja samhliða Comirnaty
- Látið lækninn eða lyfjafræðing vita um öll önnur lyf sem eru notuð, hafa nýlega verið notuð eða kynnu að verða notuð eða ef þú hefur nýlega fengið annað bóluefni.
- Meðganga og brjóstagjöf
- Við meðgöngu, brjóstagjöf, grun um þungun eða ef þungun er fyrirhuguð skal leita ráða hjá lækninum eða lyfjafræðingi áður en þú færð bóluefnið.
- Akstur og notkun véla
- Sum áhrifin af bólusetningunni sem nefnd eru í kafla 4 (Hugsanlegar aukaverkanir) geta dregið tímabundið úr hæfni þinni til aksturs eða notkunar véla. Bíddu þangað til þessi áhrif hafa gengið til baka áður en þú ekur eða notar vélar.
- Comirnaty inniheldur kalíum og natríum
- Bóluefnið inniheldur minna en 1 mmól (39 mg) af kalíum í hverjum skammti, þ.e.a.s. er sem næst kalíumlaust.
- Bóluefnið inniheldur minna en 1 mmól (23 mg) af natríum í hverjum skammti, þ.e.a.s. er sem næst natríumlaust.
- 3. Hvernig Comirnaty er gefið
- Comirnaty er gefið eftir þynningu sem 0,3 ml inndæling í vöðva í upphandleggnum.
- Þú færð 2 inndælingar.
- Ráðlagt er að fá seinni skammtinn af sama bóluefninu 3 vikum eftir fyrri skammtinn til að ljúka bólusetningunni.
- Leitið til læknisins, lyfjafræðings eða hjúkrunarfræðingsins ef þörf er á frekari upplýsingum um notkun Comirnaty.
- 4. Hugsanlegar aukaverkanir
- Eins og við á um öll lyf getur Comirnaty valdið aukaverkunum en það gerist þó ekki hjá öllum.
- Mjög algengar aukaverkanir: geta komið fyrir hjá meira en 1 af hverjum 10 einstaklingum
- • stungustaður: verkur, bólga
- • þreyta
- • höfuðverkur
- • vöðvaverkir
- • kuldahrollur
- • liðverkir
- • niðurgangur
- • hiti
Sumar þessara aukaverkana voru örlítið algengari hjá unglingum á aldrinum 12 til 15 ára en hjá fullorðnum.
Algengar aukaverkanir: geta komið fyrir hjá allt að 1 af hverjum 10 einstaklingum
- • roði á stungustað
- • ógleði
- • uppköst
- Sjaldgæfar aukaverkanir: geta komið fyrir hjá allt að 1 af hverjum 100 einstaklingum
- • stækkaðir eitlar
- • vanlíðan
- • verkur í handlegg
- • svefnleysi
- • kláði á stungustað
- • ofnæmisviðbrögð á borð við útbrot eða kláða
- Mjög sjaldgæfar aukaverkanir: geta komið fyrir hjá allt að 1 af hverjum 1 000 einstaklingum
- • skammvinn lömun í annarri hlið andlitsins
- • ofnæmisviðbrögð á borð við ofsakláða eða þrota í andliti
- Tíðni ekki þekkt (ekki hægt að áætla tíðni út frá fyrirliggjandi gögnum)
- • alvarleg ofnæmisviðbrögð
- • bólga í hjartavöðvanum eða bólga í gollurshúsi umhverfis hjarta sem geta leitt til mæði, hjartsláttarónota eða brjóstverks
- • mikil bólga í bólusettum útlim
- • bólga í andliti (bólga í andliti getur komið fram hjá sjúklingum sem hafa fengið inndælingar með fegrunarefnum í andlit)
- Tilkynning aukaverkana
Látið lækninn, lyfjafræðing eða hjúkrunarfræðinginn vita um allar aukaverkanir.
Þetta gildir einnig um aukaverkanir sem ekki er minnst á í þessum fylgiseðli.
Einnig er hægt að tilkynna aukaverkanir beint samkvæmt fyrirkomulagi sem gildir í hverju landi fyrir sig, sjá Appendix V og láta lotunúmer fylgja ef það er til staðar.
Með því að tilkynna aukaverkanir er hægt að hjálpa til við að auka upplýsingar um öryggi lyfsins.
5. Hvernig geyma á Comirnaty
Geymið lyfið þar sem börn hvorki ná til né sjá.
Eftirfarandi upplýsingar um geymslu, fyrningu, notkun og meðhöndlun eru ætlaðar heilbrigðisstarfsmönnum.
Ekki skal nota lyfið eftir fyrningardagsetningu sem tilgreind er á öskjunni og merkimiðanum á eftir EXP. Fyrningardagsetning er síðasti dagur mánaðarins sem þar kemur fram.
Geymið í frysti við -90°C til -60°C. Meðan á 6 mánaða geymslutíma stendur má geyma og flytja óopnuð hettuglös við -25°C til -15°C á samfelldu tímabili í allt að 2 vikur og geyma svo aftur við -90ºC til -60ºC.
Geymið í upprunalegum umbúðum til varnar gegn ljósi.
Flutningur frosinna hettuglasa sem geymd eru við ofurlágt hitastig (< -60°C)
• Eftir að lokaðir bakkar sem innihalda 195 hettuglös hafa verið fjarlægðir úr frystigeymslu með ofurlágu hitastigi (< -60°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 5 mínútur.
• Eftir að opnir bakkar eða bakkar sem innihalda færri en 195 hettuglös hafa verið fjarlægðir úr frystigeymslu með ofurlágu hitastigi (< -60°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 3 mínútur.
• Eftir að bökkum með hettuglösum hefur aftur verið komið fyrir í frystigeymslu eftir að hafa verið við hita sem nemur allt að 25°C, þurfa þeir að vera í frystigeymslunni í að minnsta kosti 2 klukkustundir áður en hægt er að fjarlægja þá aftur.
Flutningur frosinna hettuglasa sem geymd eru við -25°C til -15°C
• Eftir að lokaðir bakkar sem innihalda 195 hettuglös hafa verið fjarlægðir úr frystigeymslu (-25°C til -15°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 3 mínútur.
• Eftir að opnir bakkar eða bakkar sem innihalda færri en 195 hettuglös hafa verið fjarlægðir úr frystigeymslu (-25°C til -15°C) mega þeir vera við hita sem nemur allt að 25°C í allt að 1 mínútu.
Eftir að hettuglas er fjarlægt úr bakkanum skal þíða það fyrir notkun.
Eftir þíðingu á að þynna bóluefnið og nota það tafarlaust. Engu að síður hafa gögn um stöðugleika við notkun sýnt fram á að eftir að óþynnta bóluefnið hefur verið tekið úr frysti, má geyma það í allt að 1 mánuð við 2°C til 8°C. Af þessum 1 mánaðar geymslutíma við 2°C til 8°C má nota allt að 12 klukkustundir til að flytja bóluefnið. Fyrir notkun má geyma óopnað bóluefni allt að 2 klukkustundir við hitastig allt að 30°C.
Eftir þynningu á að geyma og flytja bóluefnið við 2°C til 30°C og nota innan 6 klukkustunda. Fargið öllu ónotuðu bóluefni.
Þegar hettuglösin hafa verið tekin úr frysti og þynnt, skal merkja þau með nýrri fyrningardagsetningu og -tíma. Ekki má frysta bóluefnið á ný eftir þíðingu.
Ekki má nota bóluefnið ef vart verður við agnir í þynntu lausninni eða litabreytingu.
Ekki má skola lyfjum niður í frárennslislagnir eða fleygja þeim með heimilissorpi. Leitið ráða í apóteki um hvernig heppilegast er að farga lyfjum sem hætt er að nota. Markmiðið er að vernda umhverfið.
6. Pakkningar og aðrar upplýsingar
Comirnaty inniheldur
- Virka innihaldsefnið er COVID-19 mRNA bóluefni.
- Eftir þynningu inniheldur hettuglasið 6 skammta sem hver er 0,3 ml og inniheldur 30 míkrógrömm af mRNA.
- Önnur innihaldsefni eru: − ((4-hýdroxýbútýl)azanedíýl)bis(hexan-6,1-díýl)bis(2-hexýldekanóat) (ALC-0315)
- 2-[(pólýetýlen glýkól)-2000]-N,N-dítetradekýlasetamíð (ALC-0159)
- − 1,2-Dísteróýl-sn-glýseró-3-fosfókólín (DSPC)
- kólesteról
- − kalíumklóríð
- − kalíumtvívetnisfosfat
- − natríumklóríð
- − tvínatríumfosfat tvíhýdrat
- − súkrósi
- − vatn fyrir stungulyf
Lýsing á útliti Comirnaty og pakkningastærðir
- Bóluefnið er hvít eða beinhvít ördreifa (pH: 6,9 – 7,9) í fjölskammta hettuglasi með 6 skömmtum í 2 ml glæru hettuglasi (gler af gerð I) með gúmmítappa og smelluloki úr plasti með álinnsigli.
- Pakkningastærð: 195 hettuglös
Markaðsleyfishafi
- BioNTech Manufacturing GmbH
- An der Goldgrube 12
- 55131 Mainz
- Þýskaland
- sími: +49 6131 9084-
- bréfasími: +49 6131 9084-2121
- service@biontech.de
Framleiðendur
- BioNTech Manufacturing GmbH
- Kupferbergterrasse 17 – 19
- 55116 Mainz
- Þýskaland
- Pfizer Manufacturing Belgium NV
- Rijksweg 12
- 2870 Puurs
- Belgía
|
Hafið samband við fulltrúa markaðsleyfishafa á hverjum stað ef óskað er upplýsinga um lyfið: België/Belgique/Belgien Luxembourg/Luxemburg Pfizer S.A./N.V. Tél/Tel: +32 (0)2 554 62 11 |
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. +370 52 51 4000 |
|
България Пфайзер Люксембург САРЛ, Клон България Teл: +359 2 970 4333 |
Magyarország Pfizer Kft Tel: +36 1 488 3700 |
|
Česká republika Pfizer, spol. s r.o. Tel: +420 283 004 111 |
Malta Vivian Corporation Ltd. Tel: +35621 344610 |
|
Danmark Pfizer ApS Tlf: +45 44 201 100 |
Norge Pfizer AS Tlf: +47 67 526 100 |
|
Deutschland BioNTech Manufacturing GmbH Tel: +49 6131 90840 |
Nederland Pfizer BV Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel: +372 666 7500 |
Österreich Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0 |
|
Τηλ.: +30 210 6785 800 |
Polska Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00 |
|
España Pfizer, S.L. Tel:+34914909900 |
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500 |
|
France Pfizer Tél +33 1 58 07 34 40 |
România Pfizer Romania S.R.L Tel: +40 (0) 21 207 28 00 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: +385 1 3908 777 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: +386 (0) 1 52 11 400 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel: +421 2 3355 5500 |
|
Ísland Icepharma hf Simi: +354 540 8000 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 430 040 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Sverige Pfizer AB Tel: +46 (0)8 550 520 00 |
|
Κύπρος Pfizer Ελλάς Α.Ε. (Cyprus Branch) Tηλ: +357 22 817690 |
United Kingdom (Northern Ireland) Pfizer Limited Tel: +44 (0) 1304 616161 |
|
Latvija Pfizer Luxembourg SARL filiāle Latvijā Tel.: +371 670 35 775 |




Um höfund

- ✞༺(((( Ⓒilla ℜągnąℜṧ )))༻♚༺ BA Classical Art Historian || MA Culture & Media || Tourism & Sales Management || Web Design || Photo & Videographer for Tourism Magasins ༻
Síðustu færslur
 PROTECT THE CHILDREN23. nóvember, 2024BARNAMÁLARÁÐSTEFNAN 2024
PROTECT THE CHILDREN23. nóvember, 2024BARNAMÁLARÁÐSTEFNAN 2024 MANNRÉTTINDI19. nóvember, 2024MENNTASPJALL VALGERÐAR SNÆLAND JÓNSDÓTTUR
MANNRÉTTINDI19. nóvember, 2024MENNTASPJALL VALGERÐAR SNÆLAND JÓNSDÓTTUR MANNRÉTTINDI9. ágúst, 2024Lög um borgaralega handtöku voru felld úr gildi árið 2008
MANNRÉTTINDI9. ágúst, 2024Lög um borgaralega handtöku voru felld úr gildi árið 2008 Sameinuðu Þjóðirnar28. desember, 2023Lestrarefni Menntamálastofnunar: Varúð hér býr vampíra – auðlesin sögubók á léttu máli og bókinni fylgir verkefnahefti sem hægt er að vinna samhliða lestri.
Sameinuðu Þjóðirnar28. desember, 2023Lestrarefni Menntamálastofnunar: Varúð hér býr vampíra – auðlesin sögubók á léttu máli og bókinni fylgir verkefnahefti sem hægt er að vinna samhliða lestri.